
[ad_1]
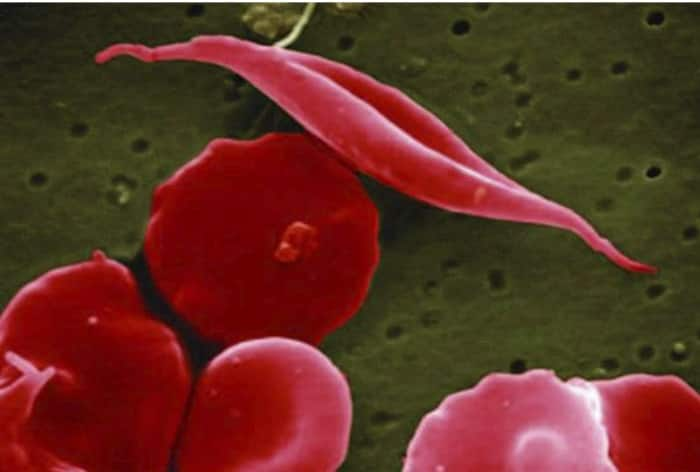
Sickle cell disease (SCD) is a painful, inherited blood disorder in which the body makes flawed, sickle-shaped hemoglobin, impairing the ability of red blood cells to properly carry oxygen to the body’s tissues.
Washington: The U.S. Food and Drug Administration (FDA) on Friday approved two gene therapies for sickle cell disease, making one of them the first treatment in the United States based on the Nobel Prize-winning CRISPR gene editing technology. The FDA approved Lyfgenia and a separate treatment called Casgevy from the patients’ own blood stem cells and were approved for people aged 12 and older. Additionally, one of these therapies, Casgevy, is the first FDA-approved treatment to utilize a type of novel genome editing technology, signaling an innovative advancement in the field of gene therapy.
The therapy can be directed to cut DNA in targeted areas, enabling the ability to accurately remove, add or replace DNA where it was cut.
Current treatments include medications and blood transfusions. The only permanent solution is a bone marrow transplant, which must come from a closely matched donor and brings a risk of rejection.
What Is Sickle Cell Disease?
Sickle cell disease (SCD) is a painful, inherited blood disorder in which the body makes flawed, sickle-shaped hemoglobin, impairing the ability of red blood cells to properly carry oxygen to the body’s tissues.
The disease, which can be debilitating and lead to premature death, affects an estimated 100,000 people in the United States, most of whom are Black.
The primary problem in sickle cell disease is a mutation in hemoglobin, a protein found in red blood cells that delivers oxygen to the body’s tissues. This mutation causes red blood cells to develop a crescent or “sickle” shape. These sickled red blood cells restrict the flow in blood vessels and limit oxygen delivery to the body’s tissues, leading to severe pain and organ damage called vaso-occlusive events (VOEs) or vaso-occlusive crises (VOCs). The recurrence of these events or crises can lead to life-threatening disabilities and/or early death.
“Sickle cell disease is a rare, debilitating and life-threatening blood disorder with significant unmet need, and we are excited to advance the field especially for individuals whose lives have been severely disrupted by the disease by approving two cell-based gene therapies today,” said Nicole Verdun, M.D., director of the Office of Therapeutic Products within the FDA’s Center for Biologics Evaluation and Research. “Gene therapy holds the promise of delivering more targeted and effective treatments, especially for individuals with rare diseases where the current treatment options are limited.”
Treatment
No donor is required for the gene therapies, which permanently change DNA in the patient’s blood cells. The goal of the Vertex therapy, called Casgevy, is to help the body go back to producing a fetal form of hemoglobin that’s present at birth — it’s the adult form that’s defective in people with sickle cell disease. CRISPR is used to knock out a gene in stem cells collected from the patient.
Lyfgenia, aims to add copies of a modified gene, which helps red blood cells produce “anti-sickling” hemoglobin that prevents or reverses misshapen cells.
When patients get the treatments, stem cells are removed from their blood and sent to a lab. Before getting the altered cells back, they must undergo chemotherapy. The process requires at least two hospitalizations, one lasting four to six weeks.
The FDA’s approval is the first for Bluebird’s treatment; Vertex has been previously authorized in Britain and Bahrain.
Data Supporting Casgevy
The safety and effectiveness of Casgevy were evaluated in an ongoing single-arm, multi-center trial in adult and adolescent patients with SCD. Patients had a history of at least two protocol-defined severe VOCs during each of the two years prior to screening. The primary efficacy outcome was freedom from severe VOC episodes for at least 12 consecutive months during the 24-month follow-up period. A total of 44 patients were treated with Casgevy. Of the 31 patients with sufficient follow-up time to be evaluable, 29 (93.5%) achieved this outcome. All treated patients achieved successful engraftment with no patients experiencing graft failure or graft rejection.
The most common side effects were low levels of platelets and white blood cells, mouth sores, nausea, musculoskeletal pain, abdominal pain, vomiting, febrile neutropenia (fever and low white blood cell count), headache and itching.
Data Supporting Lyfgenia
The safety and effectiveness of Lyfgenia is based on the analysis of data from a single-arm, 24-month multicenter study in patients with sickle cell disease and history of VOEs between the ages of 12- and 50- years old. Effectiveness was evaluated based on complete resolution of VOEs (VOE-CR) between 6 and 18 months after infusion with Lyfgenia. Twenty-eight (88%) of 32 patients achieved VOE-CR during this time period.
The most common side effects included stomatitis (mouth sores of the lips, mouth, and throat), low levels of platelets, white blood cells, and red blood cells, and febrile neutropenia (fever and low white blood cell count), consistent with chemotherapy and underlying disease.
Hematologic malignancy (blood cancer) has occurred in patients treated with Lyfgenia. A black box warning is included in the label for Lyfgenia with information regarding this risk. Patients receiving this product should have lifelong monitoring for these malignancies.
[ad_2]